胆囊癌是胆道最常见的恶性肿瘤,占胆道恶性肿瘤的80%~95%[1]。由于症状不明显或者不典型,这种疾病在发现时往往已经进展到了晚期,很多患者失去了根治性手术的机会,导致预后极差,5年生存率不足5%[2]。因此早期诊断对于改善患者预后至关重要。其中肿瘤标志物的发现是最具潜力的研究方向[3]。胆汁是由肝脏产生并经过胆道排入肠道的一种体液。胆汁的成分非常复杂,主要有胆汁酸、胆固醇、磷脂、胆红素、蛋白质和无机盐离子等。其中,蛋白质约占胆汁干重的5%~7%[4-7]。胆汁中可能含有与胆囊癌相关的蛋白质,它们局部浓度高,背景干扰小,因此胆汁蛋白质组学的研究对于发现胆囊癌相关的潜在肿瘤标志物具有重要价值。然而由于有众多杂质的干扰,胆汁蛋白质的研究比其他体液更加困难。目前文献报道的胆汁蛋白质浓度差异很大。究其原因,除了个体差异外,胆汁中复杂的成分对目前蛋白质研究方法的干扰也是重要因素。定量研究之前对其他杂质成分的去除很大程度上决定了研究的质量。因此,胆汁蛋白质的研究直到21世纪初仍然比较有限,而且局限于一些高丰度蛋白,比如血浆蛋白、黏蛋白等[8]。传统的胆汁蛋白质组学的研究方法以双向电泳为主,但是双向电泳图谱的分辨率较低[9-10],很可能漏掉某些具有重要意义的蛋白质。近年来,随着蛋白质研究技术的不断进展,体液的蛋白质组学研究取得了快速发展,尤其是同位素标记相对和绝对定量(isobaric tags for relative and absolute quantification,iTRAQ),Lable-free等一系列高通量蛋白质组学的研究方法的应用,大大推进了胆汁蛋白质组学的研究进展。上世纪80年代胆汁中发现的蛋白质不超过50种[7,11],而截至目前,胆汁中发现的蛋白质种类已经超过了3 000种[12]。然而对胆汁来讲,其复杂的成分仍然是阻碍其蛋白质组学研究的一大障碍,胆汁蛋白质的分离和纯化也是学术界不断探索的目标。在本研究中,我们探索建立了一套新的胆汁蛋白质分离纯化的方法,可以获得可靠的胆汁蛋白质样品。利用高通量的蛋白质组学研究方法iTRAQ进行蛋白质的鉴定分析,克服了二维电泳不能检出极酸、极碱、低丰度蛋白质等缺点,使得蛋白质的检出种类比传统方法大大提高。
1 材料与方法
1.1 材料
本研究3例胆囊结石患者胆囊胆汁取自第四军医大学唐都医院普外科行取石保胆术的患者,分别记为S1、S2、S3。3例胆囊癌胆汁取自行胆囊癌根治术的患者,分别记为C1、C2、C3。患者基本信息见表1。胆汁均于术中采取无菌程序抽取。抽取后立即放冰上转送至–80 ℃冰箱冻存备用。所有患者无急性胆囊炎发作,所抽取胆汁无脓、血污染。
表1 患者基本特征
Table 1 General profiles of the patients
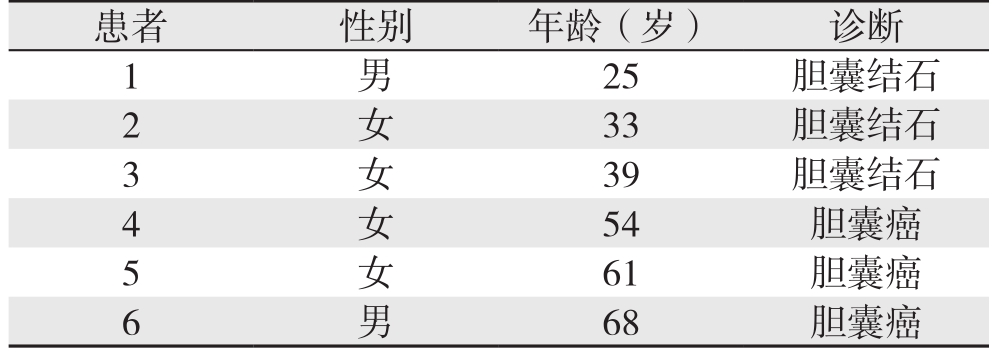
患者 性别 年龄(岁) 诊断1男25 胆囊结石2女33 胆囊结石3 39 胆囊结石4女54 胆囊癌女5 61 胆囊癌6男68 胆囊癌女
1.2 试剂和仪器
Cocktail,乙二胺四乙酸(EDTA),四甲基二乙胺(TEMED),二硫苏糖醇(DTT),甘油购于Amersco公司;胰酶(Trypsin)购于Promega公司;苯甲基磺酰氟(PMSF),三羟甲基氨基甲烷(Tris),十二烷基磺酸钠(SDS)购于BBI公司;四乙基溴化铵(TEAB),考马斯亮蓝,溴酚蓝购于Fluka公司;丙酮,异丙醇,iTRAQ标签试剂购于Fisher公司;碘乙酰胺(IAM),牛血清白蛋白(BSA)购于Sigma公司;其余试剂为国产分析纯。低温高速离心机,酶标仪为Sigma公司产品;电泳仪,凝胶扫描仪Image Scanner,图像分析软件Decyder V5.0均为Amersham公司产品;液相色谱仪LC-20AD和LC-20AB为岛津公司产品;串联质谱仪Q-Exactive为Thermo Fisher Scientific公司产品。
1.3 胆汁蛋白质的提取和定量
溶解样品,每个胆汁样品取1 mL进行提取实验;16 000 r/min 4 ℃离心15 min,取上清,加终浓度为1 mmol/L的cocktail、2 mmol/L的EDTA,混匀,置于冰上5 min后,加入终浓度10 mmol/L DTT,冰浴超声5 min;16 000 r/min 4 ℃离心15 min,取上清,将样品转置10mL离心管,加5倍冷丙酮,加入终浓度10 mmol/L DTT,-20℃沉淀蛋白2 h;15 000 r/min 4 ℃离心15 min,弃上清;加5倍冷丙酮,15 000 r/min 4 ℃离心15 min,弃上清,重复3次至上清无色;将沉淀转入2 mL离心管中,加1 mL冷丙酮,加入终浓度10 mmol/L DTT,-20 ℃沉淀蛋白2 h;25 000 r/min 4 ℃离心15 min,弃上清;风干沉淀,加适量裂解液(有SDS),终浓度为1 mmol/L的PMSF、2 mmol/L的 EDTA,混匀,置于冰上5 min后,加入终浓度10 mmol/L DTT;冰浴超声5 min;25 000 r/min 4 ℃离心15 min,将上清转入新的2 mL离心管中;加终浓度10 mmol/L DTT(蛋白液体积的1%),56 ℃水浴 1 h;冷却至室温,加终浓度55 mmol/L IAM(蛋白液体积的1/10),暗室放置45 min;5倍体积冷丙酮沉淀样品2 h,25 000 r/min 4 ℃离心15 min,弃上清;风干沉淀,加适量裂解液(无SDS);加1颗5 mm磁珠,采用Tissue lyser仪器促进蛋白质溶解(time=120 s、功率=40 Hz/s),25 000 r/min 4 ℃离心15 min,上清即为蛋白质溶液。
用Bradford法[13]对提取的蛋白质样品进行定量,测量3次取平均值。
1.4 SDS-PAGE和蛋白酶解
每样取20 μg蛋白溶液加入适量上样缓冲液混匀后95 ℃加热5 min,25 000 r/min离心5 min,取上清点入10% SDS聚丙烯酰胺凝胶的点样孔中。120 V恒压电泳120 min;电泳结束后,考马斯亮蓝染色2 h,之后加入适量脱色液(40%乙醇,10%乙酸)置于摇床脱色液3次,每次30 min。
每个样品取100 μg蛋白溶液,按蛋白:酶=40:1的比例加入Trypsin酶2.5 μg,37 ℃酶解4 h。按上述比例再补加Trypsin 1次,37 ℃继续酶解8 h。酶解的肽段利用Strata X柱进行除盐,真空抽干。
1.5 肽段标记
取出iTRAQ标签试剂,待试剂恢复至室温后,每管试剂加入50 μL异丙醇,涡旋震荡后低速离心。用0.5 mol/L TEAB溶解肽段样品,并加入到对应iTRAQ标签试剂中。不同样品肽段选用不同的iTRAQ标签。室温静置2 h。
1.6 肽段分离
采用岛津LC-20AB液相系统,分离柱为5 µm 4.6×250 mm Gemini C18柱对样品进行液相分离。用2 mL流动相A(5%CAN,pH9.8)复溶抽干的肽段样品并进样,以1 mL/min的流速梯度洗脱。5%流动相B(95% CAN,pH9.8)10 min,5%至35%流动相B 40 min,35%~95%流动相B 1 min,流动相B持续3 min,5%流动相B平衡10 min。在214 nm波长下监测洗脱峰并每分钟收集1个组分,结合色谱洗脱峰图合并样品得到20个组分,然后冷冻抽干。
1.7 高效液相色谱串联质谱
将抽干的肽段样品用流动相A(2%ACN,0.1%FA)复溶,20 000 r/min离心10 min后,取上清进样。通过纳升液相色谱仪进行分离。样品首先进入Trap柱富集并除盐,随后与自装C18柱串联,以300 nL/min流速分离。纳升液相分离末端直接连接质谱仪。经过液相分离的肽段通过nanoESI源离子化后进入到串联质谱仪Q-Exactive进行DDA(data-dependent acquisition)模式检测。
2 结 果
2.1 样品完整性检测
将提取的蛋白样品进行聚丙烯酰胺凝胶电泳和考马斯亮蓝染色后,胶图条带丰富、清晰,无明显降解和弥散,证明样品完整性良好(图1)。

图1 胆汁蛋白质样品聚丙烯酰胺凝胶电泳
Figure 1 SDS-PAGE of bile protein samples
2.2 样品浓度检测
提取的蛋白质样品经过浓度检测,达到了进行iTRAQ实验的浓度要求,所有样品蛋白总量均满足3次以上实验的需求(表2)。
表2 胆汁蛋白质样品浓度检测
Table 2 Detection of concentrations of the bile protein samples

注:1)一等样品指的是在条带合格的情况下,蛋白总量满足3次或者3次以上实验需求
Note: 1) First-class sample referring to a sample with total protein amount meeting the requirements for three or more experiments

2.3 肽段标记情况
来自每一个胆汁标本的蛋白质样品用一个标签标记,具体如表3所示。所有样品标记成功率均在85%以上,达到了iTRAQ实验所要求的标记成功率。
表3 蛋白质样品肽段标记结果
Table 3 Peptide labeling results of protein samples
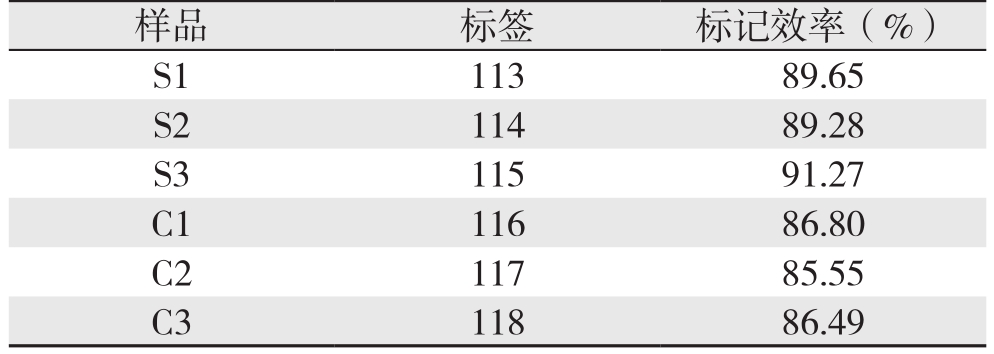
样品 标签 标记效率(%)S1 113 89.65 S2 114 89.28 S3 115 91.27 C1 116 86.80 C2 117 85.55 C3 118 86.49
2.4 蛋白质鉴定结果
经过质谱鉴定,在“1%FDR”过滤标准下,一共鉴定到1 323个蛋白。差异蛋白以变化倍数>1.5和P<0.05两个条件筛选。经筛选,胆囊癌患者胆汁与胆囊结石患者胆汁相比,173个蛋白质表达明显上调,345个蛋白质表达明显下调。单个样本间蛋白质差异情况如表4所示。对这些差异蛋白质进行进一步的生物信息学分析和验证将有可能筛选出具有诊断或预后判断意义的关键蛋白质。
表4 单个样本间差异蛋白数列表
Table 4 List of differentially expressed proteins among single
samples
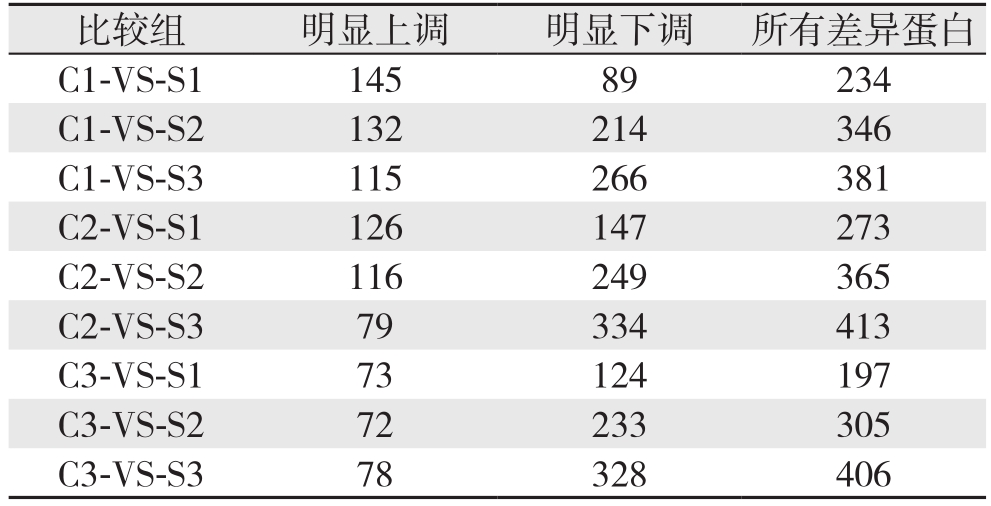
比较组 明显上调 明显下调 所有差异蛋白C1-VS-S1 145 89 234 C1-VS-S2 132 214 346 C1-VS-S3 115 266 381 C2-VS-S1 126 147 273 C2-VS-S2 116 249 365 C2-VS-S3 79 334 413 C3-VS-S1 73 124 197 C3-VS-S2 72 233 305 C3-VS-S3 78 328 406
3 讨 论
蛋白质组指一个细胞或一个组织基因组所表达的全部蛋白质,从这一水平上去探索生命现象和规律已成为研究生命科学的重要手段。经过20几年的发展,定量蛋白质组学研究方法也不断成熟,为蛋白质差异表达分析提供了更可靠、动态范围更广的研究手段,也使得生物标志物的发现成为了蛋白质组学研究的重点。目前,体液蛋白质组学的研究已经成为了发现肿瘤标志物最具潜力的研究方向[14-15]。由于胆汁与胆道疾病的发病部位长期直接接触,病变组织会通过多种机制将疾病相关的蛋白质释放到胆汁中,例如死亡或受损细胞脱落或者直接分泌。相较于血液上万种蛋白质的规模,胆汁蛋白质局部浓度高,背景干扰小,与病变部位接触更直接。加之释放到血液中的蛋白质已经被严重稀释,因此把胆汁作为蛋白质组学的研究对象比血液更有潜力发现重要的疾病相关蛋白质[16]。
然而胆汁的蛋白质组学研究面临两个主要的障碍。一是与血液、唾液、尿液等其他体液相比,胆汁的收集比较困难,除非通过手术或者逆行性胆胰管造影(ERCP)才可以获取。二是胆汁成分非常复杂,其中的有机化合物和胆汁酸盐等成分会严重干扰蛋白质的分离与鉴定。传统的基于等电点的分离方法(包括双向电泳)都会因为聚焦不良而受到影响。国内外一些学者利用双向电泳的方法对胆汁蛋白质的研究做过一些探索。他们对胆汁蛋白质的分离、纯化方法以及双向电泳的条件等进行了建立和优化,取得了一定的成果,但是由于技术方法固有的缺陷[17],双向电泳的分辨率并不理想,只能得到数百个蛋白质点,而酶解后鉴定到的蛋白质数量更加有限[9,18-21]。另外,蛋白质的质谱检测范围也有一定的限制。一些高丰度蛋白会压制其他低丰度蛋白的信号使其难以检出。色谱法也可能被一些杂质成分竞争性非特异性结合而干扰。国外有学者采用差速离心法提取胆汁蛋白质,进而进行高通量蛋白质组学研究,获得了较为理想的结果[22-23]。然而其实验条件过于苛刻,最高离心速度达200 000g,大多数实验室难以达到这样的实验条件。另外离心法也比较费时、费力,一次处理较多样本时工作量较大。
要克服以上困难,在进行蛋白质组的分析鉴定之前,简单、可靠的胆汁蛋白质样品的分离、纯化就显得十分重要。然而这里存在一个矛盾,即蛋白质提纯效果不好会影响鉴定及分析的效果,纯化可以去除干扰蛋白质鉴定的非蛋白物质,使得一些低丰度蛋白得到富集。但是过多的提取步骤则可能损失一部分蛋白质,使一些与疾病相关的重要蛋白质不能被鉴定到。因此如何把握二者之间的平衡就是一个值得探索的问题。本研究经过摸索,建立了一套胆汁蛋白质样品分离纯化的方法。提取的蛋白质样品经过质量检测,达到了进行下一步实验的要求,并取得了满意的效果。
另外由于蛋白质组学研究技术的飞速发展,传统的双向电泳的方法由于分辨率低等一些固有的缺陷,目前已经处于被逐渐淘汰的边缘,取而代之的是iTRAQ、Lable-free等高通量的强大蛋白质组学研究方法。iTRAQ技术利用多种同位素试剂标记蛋白多肽N末端或赖氨酸侧链基团,经高精度质谱仪串联分析,可同时比较多达8种样品之间的蛋白表达量,是近年来定量蛋白质组学常用的高通量筛选技术[24]。与传统研究方法相比,iTRAQ具有明显的技术优势,它能够检测出低丰度蛋白、强碱性蛋白以及分子量<10 kD或>200 kD的蛋白,分辨率远远高于双向电泳,而且操作方便,效率高,大大缩短了实验时间。
近年来由于研究手段的不断革新,蛋白质组学的研究又进入了一个快速发展的时期。2004年,Kristiansen等[25]利用SDS-PAGE、凝集素亲和层析、液相色谱串联质谱等手段,从一个胆管癌患者的胆汁中鉴定出了87种蛋白质。这是最早的胆汁蛋白质组学的研究。在接下来的数年中,胆汁蛋白质谱被逐渐扩大[23,26-28],目前发现胆汁蛋白质种类已经超过了3 000种,并且通过比较蛋白质组学的方法,发现了MAC-2BP、SSP411、NGAL、CEAM6、S100A2、α-1-抗胰蛋白酶等一批与恶性胆道梗阻相关的蛋白质分子[29-34]。
本研究建立了简单、可靠的胆汁蛋白质分离、纯化步骤,采用了iTRAQ的方法对分离提取的良恶性胆囊疾病的胆汁蛋白质样品进行了定量分析,鉴定出了1 323个蛋白质,比国内用传统方法鉴定出的蛋白质种类大有提高,并且实现了蛋白质的精确定量,发现了良恶性胆囊疾病胆汁蛋白质的显著差异,对于进一步的疾病标志物的筛选和验证具有重要应用价值。
参考文献
[1] Hundal R, Shaffer EA. Gallbladder cancer: epidemiology and outcome[J]. Clin Epidemiol, 2014, 6:99–109. doi: 10.2147/CLEP.S37357. e
[2] Rakić M, Patrlj L, Kopljar M, 等. 胆囊癌的临床诊疗现状与挑战[J]. 中国普通外科杂志, 2016, 25(2):157–161. doi:10.3978/j.issn.1005–6947.2016.02.001.Rakić M, Patrlj L, Kopljar M, et al. Current status and challenges in diagnosis and treatment of gallbladder cancer[J]. Chinese Journal of General Surgery, 2016, 25(2):157–161. doi:10.3978/j.issn.1005–6947.2016.02.001.
[3] 张华洋, 孔棣, 马波. 胆囊癌生物学标志物研究进展[J].中国普通外科杂志, 2014, 23(8):1112–1116. doi:10.7659/j.issn.1005–6947.2014.08.020.Zhang HY, Kong D, Ma B. Advances in biomarkers of gallbladder carcinoma[J]. Chinese Journal of General Surgery, 2014,23(8):1112–1116. doi:10.7659/j.issn.1005–6947.2014.08.020.
[4] Esteller A. Physiology of bile secretion[J]. World J Gastroenterol,2008, 14(37):5641–5649.
[5] Farina A, Dumonceau J, Lescuyer P. Proteomic analysis of human bile and potential applications for cancer diagnosis[J]. Expert Rev Proteomics, 2009, 6(3):285–301. doi: 10.1586/epr.09.12.
[6] Keulemans YC, Mok KS, de Wit LT, et al. Hepatic bile versus gallbladder bile: a comparison of protein and lipid concentration and composition in cholesterol gallstone patients[J]. Hepatology,1998, 28(1):11–16.
[7] LaRusso NF. Proteins in bile: how they get there and what they do[J]. Am J Physiol, 1984, 247(3 Pt 1):G199–205.
[8] Reuben A. Biliary proteins[J]. Hepatology, 1984, 4(5 Suppl):46S-50S.
[9] Stark M, Jörnvall H, Johansson J. Isolation and characterization of hydrophobic polypeptides in human bile[J]. Eur J Biochem, 1999,266(1):209–214.
[10] Chen B, Dong JQ, Chen YJ, et al. Two-dimensional electrophoresis for comparative proteomic analysis of human bile[J]. Hepatobiliary Pancreat Dis Int, 2007, 6(4):402–406.
[11] Mullock BM, Shaw LJ, Fitzharris B, et al. Sources of proteins in human bile[J]. Gut, 1985, 26(5):500–509.
[12] Farina A, Delhaye M, Lescuyer P, et al. Bile proteome in health and disease[J]. Compr Physiol, 2014, 4(1):91–108. doi: 10.1002/cphy.c130016.
[13] Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of proteindye binding[J]. Anal Biochem, 1976, 72:248–254.
[14] Ahn S, Simpson RJ. Body fluid proteomics: Prospects for biomarker discovery[J]. Proteomics Clin Appl, 2007, 1(9):1004–1015. doi:10.1002/prca.200700217.
[15] Gromov P, Gromova I, Olsen CJ, et al. Tumor interstitial fluid -a treasure trove of cancer biomarkers[J]. Biochim Biophys Acta,2013, 1834(11):2259–2270. doi: 10.1016/j.bbapap.2013.01.013.
[16] Farina A. Proximal fluid proteomics for the discovery of digestive cancer biomarkers[J]. Biochim Biophys Acta, 2014, 1844(5):988–1002. doi: 10.1016/j.bbapap.2013.10.011.
[17] Rabilloud T, Lelong C. Two-dimensional gel electrophoresis in proteomics: a tutorial[J]. J Proteomics, 2011, 74(10):1829–1841.doi: 10.1016/j.jprot.2011.05.040.
[18] 吕济相, 王济明, 罗诗樵, 等. 胆管癌胆汁蛋白质样品制备和双向电泳条件的优化[J]. 生物技术通报, 2009, (8):125–127.Lu JX, Wang JM, Luo SQ, et al. Optimization of Sample Preparation and Two-dimensional Gel Electrophoresis for Cholangiocarcinoma Bile Protein[J]. Biotechnology Bulletin, 2009,(8):125–127.
[19] 陈波, 唐滔, 郑建伟, 等. 胆汁双向电泳差异分析样本纯化制备方法的建立[J]. 外科理论与实践, 2007, 12(5):468–471. doi:10.3969/j.issn.1007–9610.2007.05.018.Chen B, Tang T, Zheng JW, et al. Establishment of a sample preparation strategy for two-dimensional electrophoresis of human bile[J]. Journal of Surgery Concepts & Practice, 2007, 12(5):468–471. doi:10.3969/j.issn.1007–9610.2007.05.018.
[20] 陈波, 郑建伟, 王剑明, 等. 人胆汁蛋白质组双向电泳图谱的建立与初步分析[J]. 中华肝胆外科杂志, 2007, 13(8):536–539.doi:10.3760/cma.j.issn.1007–8118.2007.08.013.Chen B, Zheng JW, Wang JM, et al. Establishment and preliminary analysis of a 2-D human biliary map[J]. Chinese Journal of Hepatobiliary Surgery, 2007, 13(8):536–539. doi:10.3760/cma.j.issn.1007–8118.2007.08.013.
[21] 黄国飞, 王济明, 罗诗樵, 等. 人胆汁双向凝胶电泳分离技术的建立与优化[J]. 中国组织工程研究与临床康复, 2009, 13(15):2937–2941. doi:10.3969/j.issn.1673–8225.2009.15.029.Huang GF, Wang JM, Luo SQ, et al. Construction and optimization of a separation technique of human bile using twodimensional gel electrophoresis[J]. Chinese Journal of Tissue Engineering Research, 2009, 13(15):2937–2941. doi:10.3969/j.issn.1673–8225.2009.15.029.
[22] Lukic N, Visentin R, Delhaye M, et al. An integrated approach for comparative proteomic analysis of human bile reveals overexpressed cancer-associated proteins in malignant biliary stenosis[J]. Biochim Biophys Acta, 2014, 1844(5):1026–1033. doi:10.1016/j.bbapap.2013.06.023.
[23] Farina A, Dumonceau J, Delhaye M, et al. A step further in the analysis of human bile proteome[J]. J Proteome Res, 2011,10(4):2047–2063. doi: 10.1021/pr200011b.
[24] Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents[J]. Mol Cell Proteomics, 2004,3(12):1154–1169.
[25] Kristiansen TZ, Bunkenborg J, Gronborg M, et al. A proteomic analysis of human bile[J]. Mol Cell Proteomics, 2004, 3(7):715–728.
[26] Zhou H, Chen B, Li RX, et al. Large-scale identification of human biliary proteins from a cholesterol stone patient using a proteomic approach[J]. Rapid Commun Mass Spectrom, 2005, 19(23):3569–3578.
[27] Farid SG, Craven RA, Peng J, et al. Shotgun proteomics of human bile in hilar cholangiocarcinoma[J]. Proteomics, 2011, 11(10):2134–2138. doi: 10.1002/pmic.201000653.
[28] Barbhuiya MA, Sahasrabuddhe NA, Pinto SM, et al.Comprehensive proteomic analysis of human bile[J]. Proteomics,2011, 11(23):4443–4453. doi: 10.1002/pmic.201100197.
[29] Koopmann J, Thuluvath PJ, Zahurak ML, et al. Mac-2-binding protein is a diagnostic marker for biliary tract carcinoma[J]. Cancer,2004, 101(7):1609–1615.
[30] Shen J, Wang W, Wu J, et al. Comparative proteomic profiling of human bile reveals SSP411 as a novel biomarker of cholangiocarcinoma[J]. PLoS One, 2012, 7(10):e47476. doi:10.1371/journal.pone.0047476.
[31] Zabron AA, Horneffer-van der Sluis VM, Wadsworth CA, et al.Elevated levels of neutrophil gelatinase-associated lipocalin in bile from patients with malignant pancreatobiliary disease[J]. Am J Gastroenterol, 2011, 106(9):1711–1717. doi: 10.1038/ajg.2011.187.
[32] Farina A, Dumonceau J, Antinori P, et al. Bile carcinoembryonic cell adhesion molecule 6 (CEAM6) as a biomarker of malignant biliary stenoses[J]. Biochim Biophys Acta, 2014, 1844(5):1018–1025. doi: 10.1016/j.bbapap.2013.06.010.
[33] Wasuworawong K, Roytrakul S, Paemanee A, et al. Comparative Proteomic Analysis of Human Cholangiocarcinoma Cell Lines:S100A2 as a Potential Candidate Protein Inducer of Invasion[J]. Dis Markers, 2015, 2015:629367. doi: 10.1155/2015/629367.
[34] Laohaviroj M, Potriquet J, Jia X, et al. A comparative proteomic analysis of bile for biomarkers of cholangiocarcinoma[J].Tumour Biol, 2017, 39(6):1010428317705764. doi:10.1177/1010428317705764.
